Researchers used genomic structural equation modeling to separate schizophrenia-specific and shared bipolar genetic risks. Schizophrenia-unique variants were linked to lower IQ, while shared variants correlated with higher education.

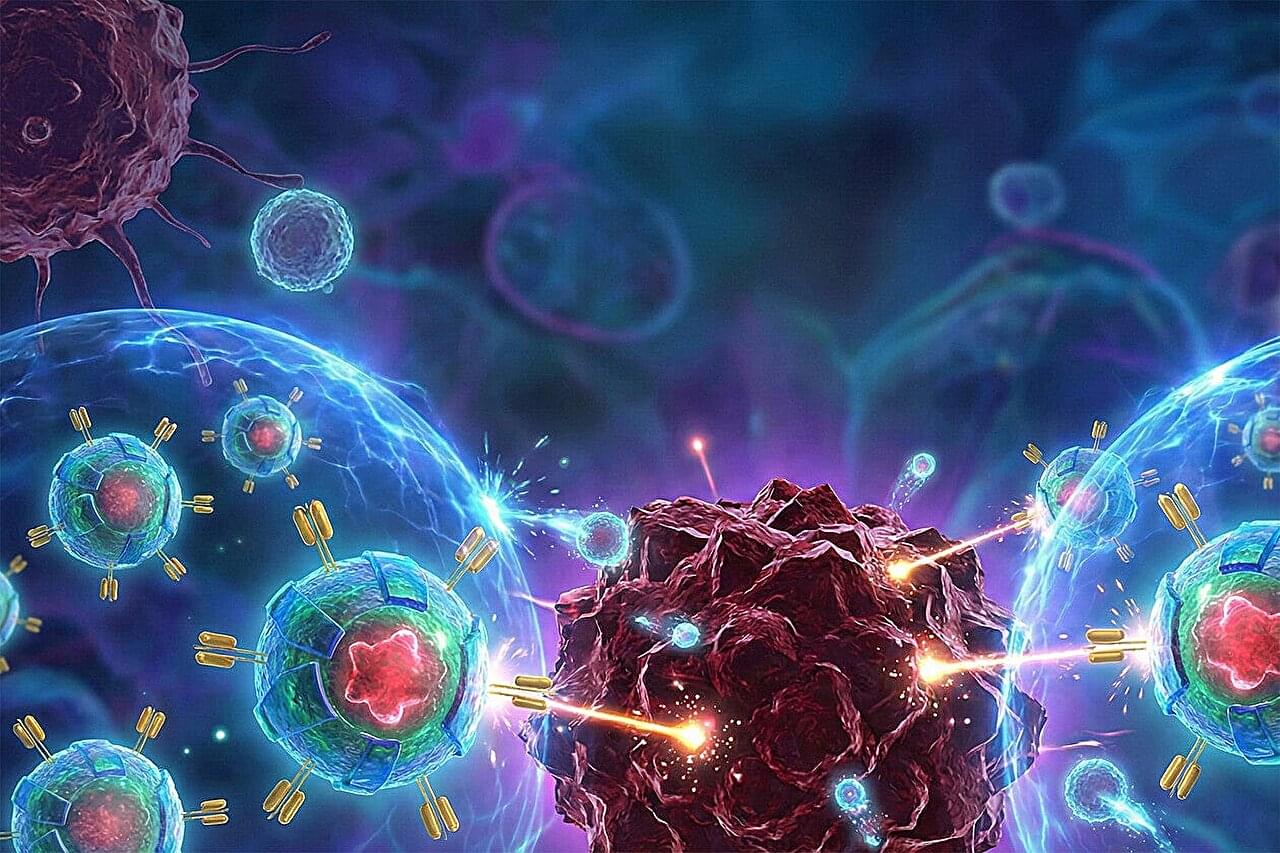
Since scientists first discovered that human immune cells could be modified to become cancer-fighting agents, they’ve been trying to engineer a cell that’s effective against solid tumors, which account for the vast majority of cancer cases. In a key advance in meeting this “holy grail challenge” in the field of cancer cell therapy, a team of Yale scientists led by geneticist Sidi Chen has revealed how immune cells can be “boosted” to target and eradicate solid tumors.
The field of cell therapy began to revolutionize cancer treatment several decades ago, when researchers pioneered the use of therapeutic cells. In this process, immune cells are removed from a patient, modified so that they can better fight cancer, and then reintroduced into the patient’s body.
Two major streams of this therapy exist: CAR-NK cell therapy, which uses a patient’s natural killer (NK) cells, and CAR-T cell therapy, which uses a patient’s T cells. In both cases, scientists genetically modify the cells to express Chimeric Antigen Receptor (CAR), a synthetic receptor that helps immune cells recognize proteins on cancer cells.
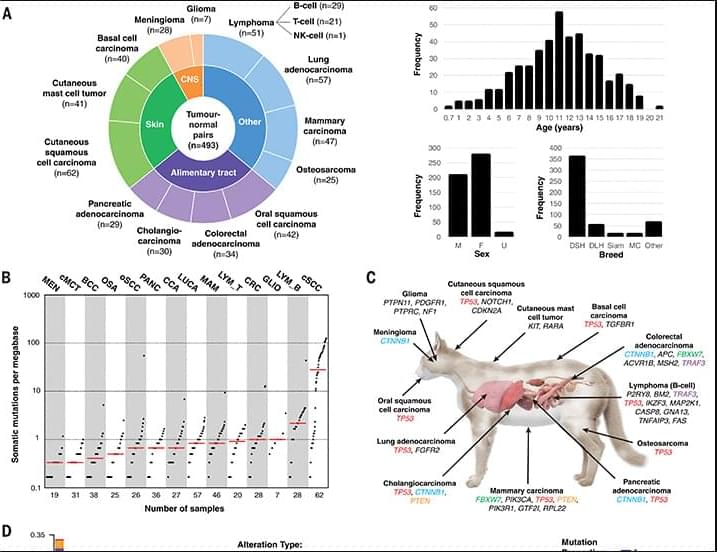
Although cancer is a common cause of death in domestic cats, little is known about the range of cancer genes in cat tumors, and how this range might compare with the oncogenome in people.
Now, researchers in Science have sequenced cancer genes in 493 samples from 13 different types of feline cancer and matched healthy control tissue, gaining a clearer picture of the cat oncogenome and comparing the genes to known cancer-causing mutations in humans.
Cancer is a common cause of morbidity and mortality in domestic cats. Because the mutational landscape of domestic cat tumors remains uncharacterized, we performed targeted sequencing of 493 feline tumor–normal tissue pairs from 13 tumor types, focusing on the feline orthologs of ~1000 human cancer genes. TP53 was the most frequently mutated gene, and the most recurrent copy number alterations were loss of PTEN or FAS or gain of MYC. By identifying 31 driver genes, mutational signatures, viral sequences, and tumor-predisposing germline variants, our study provides insight into the domestic cat oncogenome. We demonstrate key similarities with the human oncogenome, confirming the cat as a valuable model for comparative studies, and identify potentially actionable mutations, aligning with a “One Medicine” approach.

…Life Biosciences, a biotech company co-founded by Sinclair, received the FDA’s approval to begin a human trial testing its gene therapy based on the Information Theory of Aging. The gene therapy is designed to rewind the clock and restore the function of dying cells…
…Life Biosciences’ gene therapy has been under development for quite a while. In the 1990s, David Sinclair first contended that the deterioration and loss of epigenetic information—chemical tagging patterns on DNA that regulate which genes are turned on and off—plays an important role in driving aging. Sinclair subsequently dubbed this contention the Information Theory of Aging. Fast forward to the present day, and Life Biosciences has produced a gene therapy that delivers three proteins, which Sinclair’s laboratory helped establish, to reset epigenetic information to a more youthful state.
‘It’s extremely exciting,’ Sinclair told Endpoints News. ‘It’s been over 30 years to get to this point, and we’re about to learn if all of that work is going to come to fruition this year.’
The FDA has greenlighted Life Biosciences’ first human trial testing whether their gene therapy can confer a near-total rejuvenating reset of cells.
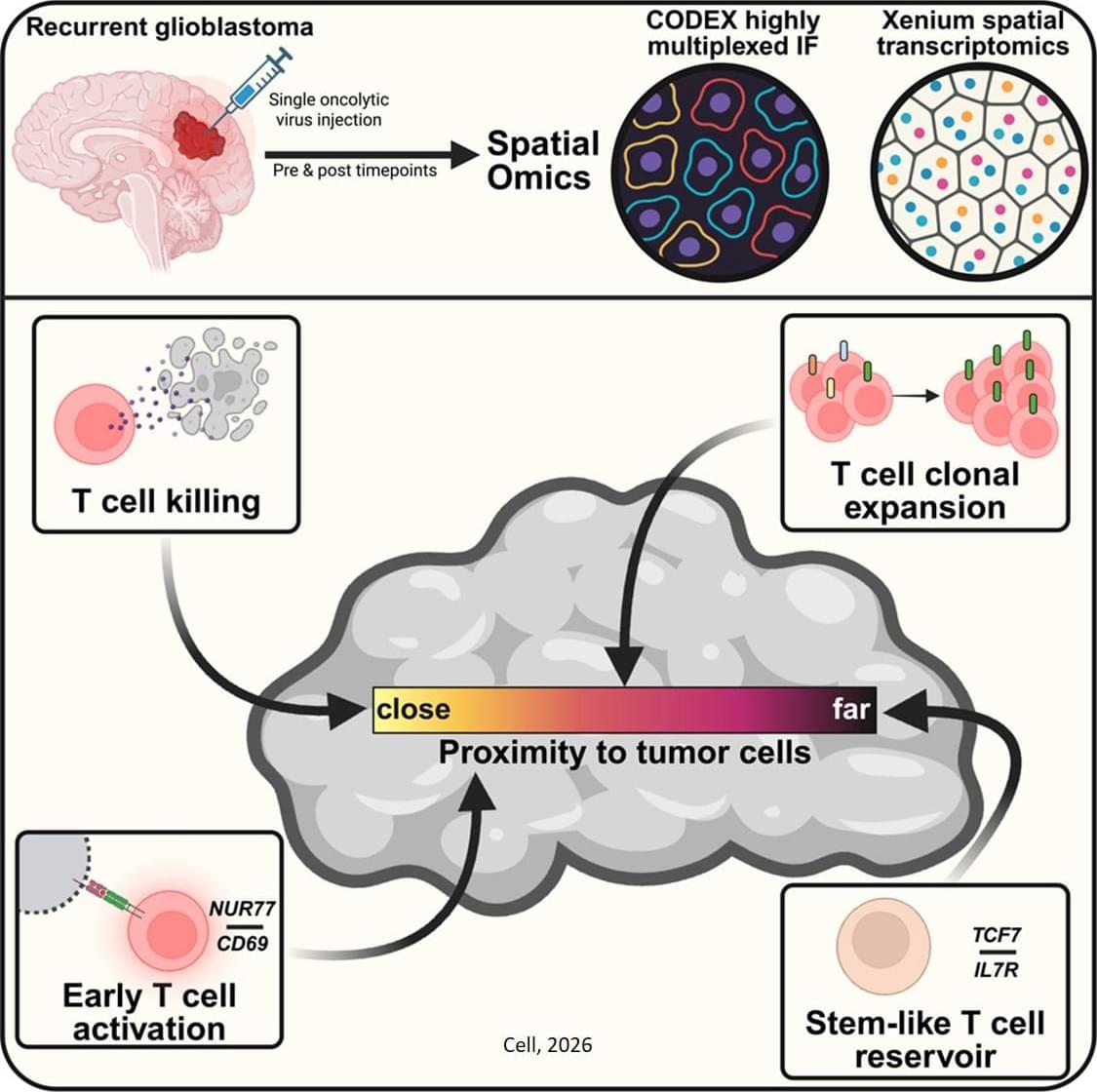
A team led by investigators has shown that a single injection of an oncolytic virus—a genetically modified virus that selectively infects and destroys cancer cells—can recruit immune cells to penetrate and persist deep within brain tumors. The research, which is published in Cell, provides details on how this therapy prolonged survival in patients with glioblastoma, the most common and malignant primary brain tumor, in a recent clinical trial.
The oncolytic virus used in the team’s trial is made from a herpes simplex virus genetically altered so it can only make copies of itself in glioblastoma cells and not normal healthy cells. The virus spreads to a glioblastoma cell, kills it, and then makes a copy of itself that spreads again to another glioblastoma cell. Infection of cells with the virus also triggers an immune response. In the phase 1 trial of 41 patients with recurrent glioblastoma, the oncolytic virus treatment extended survival compared to historically reported survival, especially among those with pre-existing viral antibodies.
In their Cell study, the investigators examined the extent of this immune response in clinical trial participants. Their analysis revealed that the treatment induced long-term infiltration of immune T cells into patients’ tumors. Closer proximity of cytotoxic T cells with dying brain tumor cells was associated with longer patient survival after treatment. The therapy also expanded pre-existing T cells in the brain. ScienceMission sciencenewshighlights.
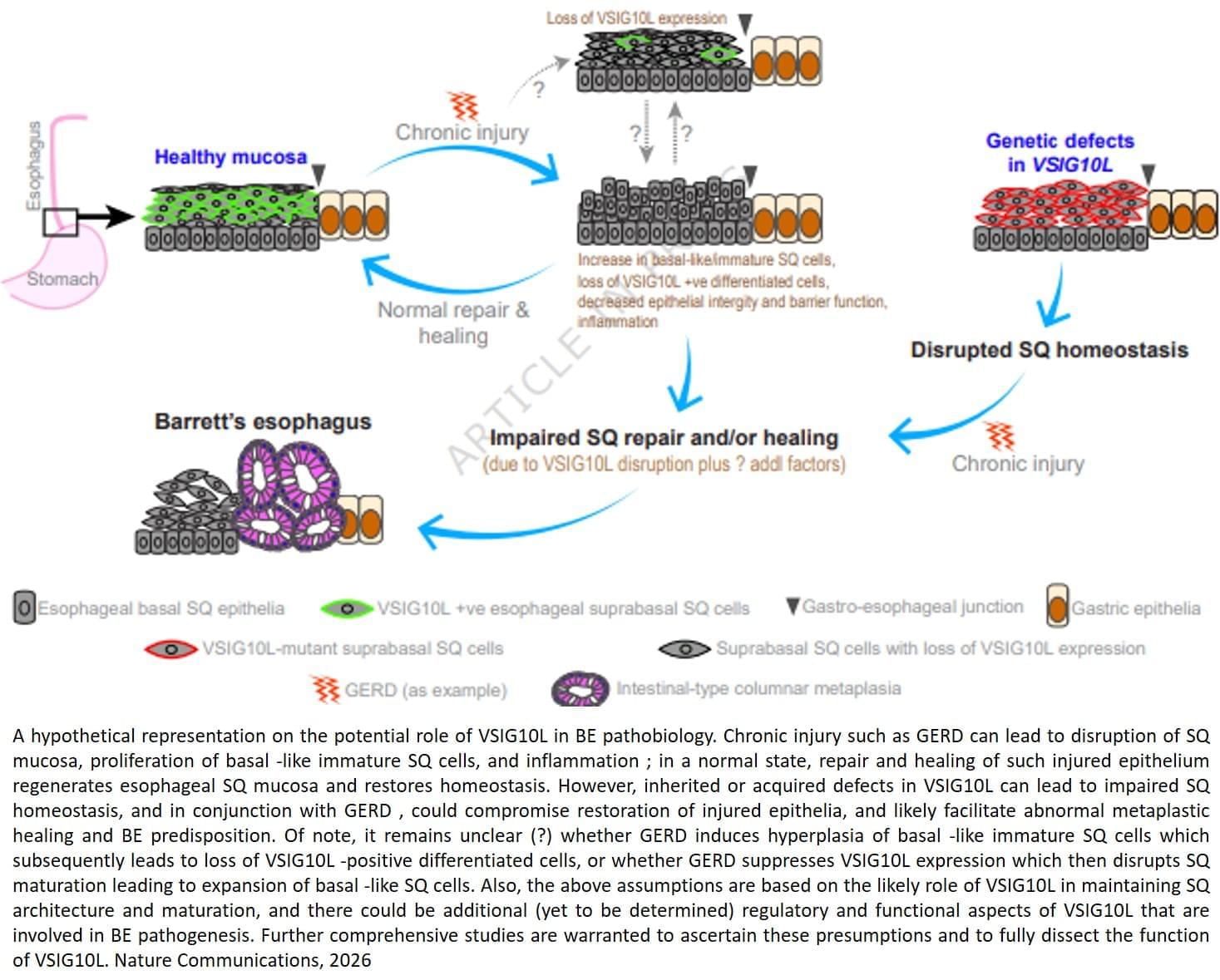
But the molecular factors responsible for the onset of Barrett’s esophagus remain poorly understood.
The findings, published in Nature Communications, combined family studies, laboratory experiments and genetically engineered mouse models to identify and understand how genetic defects contribute to disease development.
The team sequenced and analyzed genetic material of 684 people from 302 families where multiple members developed Barrett’s esophagus or esophageal cancer. They discovered that a subset of affected family members carry inherited mutations in a gene called VSIG10L.
“We found that this gene acts like a quality control system for the esophageal lining,” said the lead researcher. “When it’s defective, the cells do not mature properly and the protective barrier in the esophageal lining becomes weak, allowing stomach bile acid to cause tissue changes that enhances the risk of developing Barrett’s esophagus.”
When researchers genetically engineered mice with human-equivalent VSIG10L mutations, they found that the esophageal lining became disrupted structurally and molecularly, according to the author. The study found that when the mice were exposed to bile acid, they developed Barrett’s-like disease over time, effectively replicating the disease’s progression in humans.
These genetically engineered mice also represent the first animal model for Barrett’s esophagus based directly on human genetic predisposition to the disease, the author said.
With VSIG10L shown to be a key gene in maintaining esophageal health, family members can now be screened for genetic variants to identify those at a high-risk of developing Barrett’s esophagus or esophageal cancer. ScienceMission sciencenewshighlights.
In All Tomorrows by C. M. Kosemen, also known as Nemo Ramjet, humanity’s distant descendants are reshaped across millions of years into wildly divergent “post-human” species after being genetically engineered by the godlike alien Qu. These forms range from tiny, almost vermin-like organisms and sessile, colony-bound beings to aquatic leviathans, aerial gliders, and towering, heavily built giants as each adapted to extreme planetary environments and radically different evolutionary pressures. Some retain echoes of recognizable humanity, while others are so transformed they blur the line between animal, ecosystem, and living architecture. In this size comparison, we’ll explore the full spectrum of these post-human forms, from the smallest engineered remnants to the most massive macro-organic descendants.
Credits:
https://all-tomorrows.fandom.com/wiki/Qu.
https://speculativeevolution.fandom.com/wiki/All_Tomorrows.
FAIR-USE COPYRIGHT DISCLAIMER Copyright Disclaimer under Section 107 of the Copyright Act 1976, allowance is made for “fair use” for purposes such as criticism, commenting, news reporting, teaching, scholarship, and research. Fair use is a use permitted by copyright statute that might otherwise be infringing. Non-profit, educational or personal use tips the balance in favour of fair use. Nutbug does not own the rights to these videos and pictures. They have, in accordance with fair use, been repurposed with the intent of educating and inspiring others. However, if any content owners would like their images removed, please contact us by email [email protected]
Join us on Patreon! https://www.patreon.com/MichaelLustgartenPhD
Discount Links/Affiliates:
Blood testing (where I get the majority of my labs): https://www.ultalabtests.com/partners/michaellustgarten.
Blood testing with LifeExtension.com: https://www.anrdoezrs.net/click-101614996-15750394
At-Home Metabolomics: https://www.iollo.com?ref=michael-lustgarten.
Use Code: CONQUERAGING At Checkout.
Clearly Filtered Water Filter: https://get.aspr.app/SHoPY
Epigenetic, Telomere Testing: https://trudiagnostic.com/?irclickid=U-s3Ii2r7xyIU-LSYLyQdQ6…M0&irgwc=1

💬 Editorial: Precision adjuvant therapy for stage III ColonCancer may be enhanced through molecular profiling for ctDNA status and PIK3CA mutation, informing use of celecoxib or aspirin alongside standard treatment.
CALGB/SWOG 80,702 Alliance was a placebo-controlled randomized clinical trial (RCT) of daily celecoxib (400 mg/d vs placebo) as an adjuvant therapy to fluorouracil, leucovorin, and oxaliplatin (FOLFOX) toward improving disease-free survival (DFS) of minimal residual localized (stage III) metastatic colon cancer.1 The rationale for the trial was a preponderance of evidence from RCTs and observational studies showing that selective cyclooxygenase 2 (COX-2 or prostaglandin-endoperoxide synthase 2 [PTGS2]) inhibitors, nonsteroidal anti-inflammatory drugs (NSAIDs) such as celecoxib and rofecoxib, reduce the incidence of premalignant colorectal polyps and colorectal cancer (CRC). Although the primary trial results did not show daily celecoxib to be statistically significantly associated with improvement in DFS or overall survival (OS),1 the results raised the possibility that yet-to-be-determined subgroups may experience a significant benefit. Indeed, Nowak et al2 reported in 2024 that a significant protective effect was observed among patients with tumors harboring mutations to exons 9 or 20 of the PIK3CA gene within the subset of the Alliance trial population with available whole-exome tumor sequencing data.
The possibility for molecular selection for NSAID adjuvant therapy of CRC, specifically on the basis of PIK3CA mutation was first raised in a prospective observational study by Liao and colleagues3 in 2012 for aspirin—a less selective COX-2 inhibitor. This finding for aspirin was later corroborated with post hoc observational follow-up of the VICTOR RCT of daily rofecoxib (20 mg vs placebo),4 which, like the Alliance trial, did not demonstrate a significant protective benefit for rofecoxib among unselected patients.5 Most recently, 2 RCTs of daily low-dose aspirin, ALASSCA6 and SAKK41/13,7 showed that aspirin, among patients enrolled using molecular selection for tumor PIK3CA mutation, led to a similar survival benefit of approximately 50% compared to placebo.